Hi everyone, long-time lurker, first-time poster here (also posted it on Reddit earlier today). This is my longwinded theory regarding a possible cause and cure for androgenetic alopecia. I’m by no means claiming to be a scientist and I apologise in advance for how wordy this post is. Citations are at the bottom.
I’ve been researching androgenetic alopecia (Androgenetic Alopecia) considerably for a few years now ever since I first noticed its effect on myself. Like many, I pussy-footed around the reality of the situation, convincing myself that the higher hairline and thinning crown was stress-related Telogen Efflivium, some basic dietary deficiency or the result of too much booze and smokes. This meant that I delayed treatment with more conventional (and proven) methods for a little longer than I probably should have.
My apprehension to begin these treatments was by enlarge because of how unsatisfied I was with the widely-accepted ‘cause’ of hair loss: that, not everybody (for some reason), but a large group (for some reason) of unlucky fellows and some women develop a bizarre, localized sensitivity (for some reason) to an androgen that their own body produces naturally and that (for some reason) this hormone ‘attacks’ tens of thousands of tiny organs on a specific part of their head (… for some reason). I thought the logic was dumb and that that kind of thinking did not a cure make.
Now, the human body is by no means a perfect machine, but it didn’t sit well with me that certain genomes would evolve something as complicated and energy-consuming as hair growth only to have it inadvertently switched off by a naturally-occurring androgen. There were also other evidences that it wasn’t just a matter of androgen activity: how often people with Androgenetic Alopecia do not display more testosterone (or its more active metabolite dihydrotestosterone [DHT]) than people unaffected; that simply blocking DHT production does not regrow hair in all people; where and the pattern in which the hair is lost (and isn’t); and lastly, the irrefutable links Androgenetic Alopecia has to issues elsewhere in the body such as heart and prostate health.
So I dug deeper into other theories a little more substantial than “DHT BAD” – ideally to find one that gave some insight into the process of hair loss being something the body decides to do rather than something it has done unto it. I read through almost every major theory on the web: tension in the galea; reduced brown adipose tissue; mast cell activation, histamine or prostaglandin imbalance etc. None of them seemed outrageous, but none of them gave much reason for Androgenetic Alopecia other than some people simply lose the genetic lottery. Eventually I stumbled upon something I did like: the phenomenon of seasonal hair growth [1]. Put simply, the idea is that the scalp is like a UV-ray sponge for the body to use to produce vitamin D, and the amount of hair on the scalp can be considered a valve that closes (grows) to reduce UV exposure, or opens (sheds) to increase it.
This theory (and the various studies relating to it) appeased me because they suggested that an intentional and strategic biological hair loss mechanism exist in every human, regardless of whether they suffer from more permanent types of alopecia. It was comforting to know that even the thickest head of hair technically undergoes ‘hair loss’ for a large portion of the year. My thinking then became that some of us must get stuck in a sort of ‘perpetual winter’ state where the valve gets stuck open.
I was obviously nowhere near the first person to theorise that vitamin D played a major role in alopecia (as a quick Google search would tell you), but the cure seemed simple: if the body has a mechanism to keep itself in a desired range of circulating cholecalciferol (vitamin D3), then oral supplementation – which has been proven to increase serum cholecalciferol [2] – might make the body think it’s getting enough sun exposure to close the valve and regrow hair for good. However, a quick skim of numerous Reddit and hair loss forum discussions quickly shoot down that idea, with many members reporting no major (although some minor) improvements from vitamin D3 supplementation alone, with bloodwork confirming they are not deficient. So it would appear we’re back to square one and pointing the finger angrily at DHT again.
However, cholecalciferol in and of itself is largely useless when it comes to hair. What is important is the vitamin D Receptor (VDR). The VDR is a nuclear receptor found in almost every cell in the body – including scalp hair follicles – and plays a direct role in the cellular proliferation, function and health of hair growth [3]. To make it clear, a follicle with more activated VDR will produce thicker hair faster than a follicle with less VDR activation.
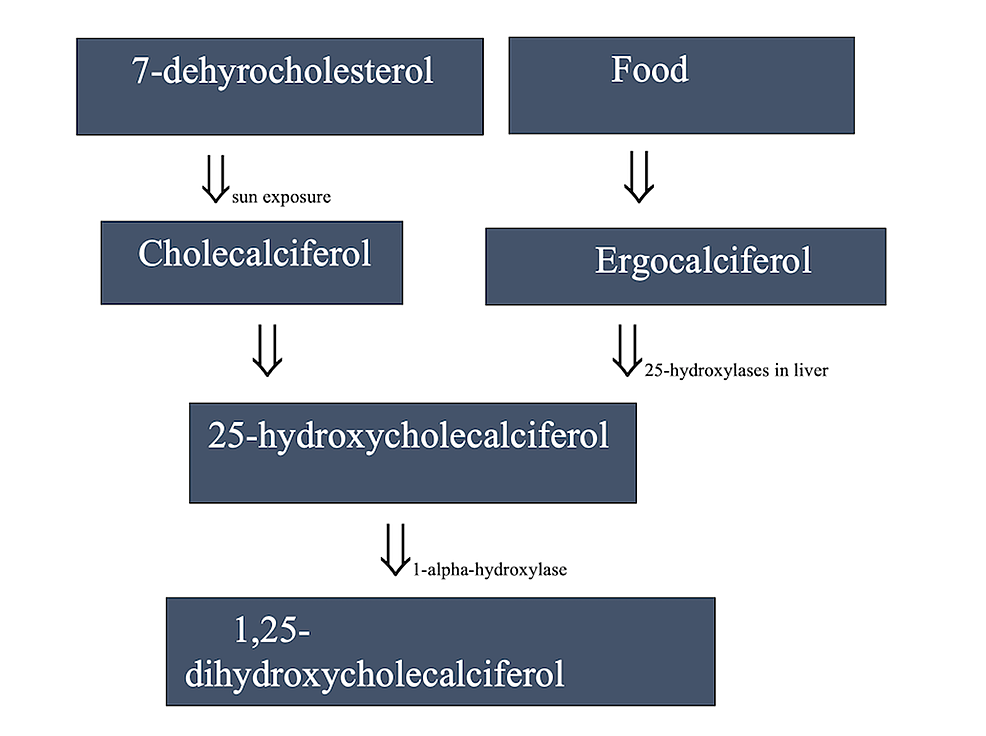
The issue with the simplicity of my initial thinking was that cholecalciferol cannot adequately activate the VDR by itself. It must first undergo conversion to calcifediol (25-hydroxyvitamin D) in the liver, and then undergo another conversion in the kidneys to its active form, 1,25-dihydroxycholecalciferol, also known as calcitriol. Calcitriol can activate the VDR and actually has such a high affinity (roughly 1000 times greater than cholecalciferol) for doing so that the VDR is sometimes referred to as the ‘calcitriol receptor’ in medical literature.
Where it gets confusing (and more than a little frustrating), is that you can have more than one blood vitamin D value that may not correlate to the other. Most doctors will test for ‘25 OH vitamin D’, also known as 25-hydroxyvitamin D, or calcifediol, as mentioned earlier. This value is good for testing for deficiency, but is more or less useless in relation to VDR activation as calcifediol cannot activate the receptor well either. Doctors can test for 1,25-dihydroxyvitamin D (calcitriol), but this will not give an insight into the body’s stores of the fat-soluble vitamin. In fact, blood calcitriol can actually spike if there is a deficiency of stored calcifediol, which further complicates the results [4].
Deciphering this means, basically, that your common bloodwork for vitamin D can come back with healthy (or even above-ideal) numbers of stored, inactive D3, but shed zero light on the circulating calcitriol metabolite that can activate the VDR. It’s also been established that the ratio at which calcitriol is converted from calcifediol (the 1,25D:25D) can vary substantially from person to person: “the ratio between calcitriol and calcifediol serum concentrations could suggest vitamin D hydroxylation efficiency.” [5]. Hydroxylation is “a chemical process that introduces a hydroxyl group (-OH) into an organic compound. In biochemistry, hydroxylation reactions are often facilitated by enzymes called hydroxylases.” [6]. What this means is that hydroxylation is what makes regular vitamin D (from sun exposure or a supplementation) able to activate the VDR and grow hair, and that it’s possible that a lowered natural capacity for vitamin D hydroxylation could result in less circulating calcitriol and thus lower systemic VDR activation.
While I couldn’t find any studies alluding to what the 1,25D:25D ratio might have been in our forefathers, it is not hard to find studies referencing vitamin D’s hydroxylation cofactors – the primary one of which being magnesium, an element that a huge proportion of the population are deficient in and one stripped from the body by modern diet and lifestyle [7]. On top of magnesium being necessary to produce calcitriol (and thus directly implicated in VDR activation), it’s also been shown that inadequate magnesium levels lead to calcification of soft tissues and that adequate levels protect against it [8]. Interestingly, and as we probably all know by now, biopsies of bald scalps have shown considerable calcification in their soft tissues compared to non-balding scalps. More interestingly, is that the negative effects of low magnesium can also be seen in the heart and prostate (in hypertension and benign prostate hyperplasia) – and that these are the organs and conditions that the only two FDA-approved medications for hair loss (minoxidil and finasteride) were originally designed to alleviate.
So, all I’ve really talked about thus far is the VDR. The glaring hole in the theory is that the internet has seen a plethora of success stories from people taking anti-androgens and minoxidil (among others that I probably won’t get into). “Hey, idiot, if the VDR is solely responsible for hair cycling, then why do these drugs that don’t have any direct action on the VDR work for so many people?” you might ask, and it’d be a damn good question. I stalled on this part of my theory for a long time but I’m glad I did, because I think this is where mine differs from others, so here it is, in a nutshell…
I believe Androgenetic Alopecia is the result of a bad ratio of VDR to androgen receptor (AR) activation and density within the body.
There seems to me to be four primary and irrefutable facts that science has concluded regarding these receptors and their relation to hair growth and Androgenetic Alopecia:
· One: activating the AR in a hair follicle (like DHT can) causes a decrease of the anagen (growth) phase and eventually a complete miniaturisation of the follicle. [9]
· Two: activating the VDR in a hair follicle (like calcitriol can) causes an increase of the angagen phase which produces a thicker, faster-growing hair. [3]
· Three: inhibiting activation of the AR in a hair follicle (like an anti-androgen can) arrests further hair loss in most people, regrows in some but does not prevent further loss in others. [10]
· Four: inhibiting activation of the VDR in a hair follicle (like low levels of calcitriol or low VDR density can) causes a decrease of the anagen phase and eventually shedding. [11]
My theory is this: I believe there exists an ideal homeostasis of VDR and AR inside every organ in the body for healthy and proper function, but most modern diets and lifestyles are throwing out this balance. It’s important to place equal significance on each of these receptors in terms of Androgenetic Alopecia. I think of the balance as a tug-of-war, with the AR wanting to turn the follicle down to zero and the VDR wanting to turn it to max. As long as the ratio of VDR:AR tilts in the VDR’s favour by even the slightest amount, you will keep your hair.
This might also allude as to why it’s exceptionally rare, but not impossible for women to develop Androgenetic Alopecia, as they have very low (but never zero) circulating androgens.
Two quite ironic cases I came across in my research show the power of each receptor in isolation. Firstly, eunuchs (boys that are castrated before puberty and don’t ever produce considerable quantities of testosterone or DHT) never lose their hair [12], and secondly, a case of a seven year old boy with hereditary vitamin D-dependent rickets type II ([VDDR-II] (a condition where the VDR is resistant to activation from calcitriol, thus experiencing almost zero activation systemically,) who suffered alopecia totalis at an age too young to have begun developing sizeable amounts of testosterone or DHT [13]. So, we have grown men who never lost a single scalp hair because of minuscule AR receptor activation, and a young boy who never grew a single scalp hair because of miniscule VDR activation.
It was all starting to make sense to me, but I still wasn’t happy with why exactly the AR – a receptor that exists naturally in the follicle – would effectively act to damage it. With further reading, I came to the conclusion that there are two primary reasons why this homeostasis is so important and why going out of it leads to almost exponential and often irreversible loss. The first of which has to do with not only the ratio of receptors themselves, but also how they interact with the other:
While there has not been extensive research into the interplay (crosstalk) of these two receptors within hair follicles, there have been numerous studies regarding how they react in other tissues – namely the prostate. Although vastly different organs, the 5-alpha-reductase type II enzyme ([5ARII], one of three responsible for converting testosterone into DHT,) is most prevalent in scalp and prostatic tissues [14]. Finasteride has only been approved by the FDA to deal with complications in these two tissues by selectively targeting the 5ARII enzyme, suggesting there may be a similar mode of suppression in each. In studies of prostate cancer, VDR activation has been shown to suppress AR expression in certain cell lines [15] while AR activation has been shown to suppress VDR activation [16] [17].
Further to the expressional crosstalk between the two receptors, there is also the matter of up/down regulation and receptor density. Cells in the prostate and scalp possess both AR and VDR (among other receptors) in varying densities. There have been numerous studies showing that: AR can be upregulated with regular activation [18] [19]; AR can be downregulated by prolonged inactivation [20]; VDR can be upregulated (and protected) by activation [21] [22]; VDR can be downregulated by deficiency [23].
For me, this explains why there are such varying severities of hair loss amongst individuals and why anti-androgens such as finasteride work for some better than others. I believe the issue and cure is not simply reducing AR activation, but reducing it to a point where it is exceeded by VDR activation. Finasteride reduces scalp DHT by roughly 64% at a 1mg daily dosage [24]. As a very rudimentary example, say you have some hair follicles with 20 active AR but only 12 active VDR. A 1mg dose of finasteride should (in theory) reduce that number of activated AR by 64% to around 7.2, creating a surplus of VDR activation and thus regrowing hair. This is what I believe happens in follicles that respond well to anti-androgen treatment.
But say, due to VDR downregulation and AR upregulation (which often go hand-in-hand), you had follicles with 30 active AR and only 10 active VDR. That same finasteride dose will create a larger drop in AR activation than the previous example (19.2 vs 12.8), but will only bring the total activated AR down to 10.8 – still exceeding the activated VDR count and thus maintaining an environment where the VDR is under-expressed compared to the AR and hair loss continues. My theory posits a few things here: firstly, that great responders to anti-androgens do not have a major deficit in their VDR:AR to contend with; secondly, that poor responders to anti-androgens do have a major deficit in their VDR:AR that some anti-androgens are not powerful enough to correct; and thirdly, that anti-androgens such as finasteride rarely produce a complete reversal of hair loss due to differing ratios of VDR:AR across the scalp follicles, as some follicles have gone beyond the drug’s ‘saveable threshold’.
Further to this, I feel the theory also provides some explanation as to the ‘catch up hair loss’ phenomenon, whereby people who regrow hair with an anti-androgen and then end treatment often find that their hair is lost in a more severe pattern than it was pre-treatment, i.e. they lose even more hair post-treatment than they had lost before it. In these cases, I would think the healthy hair they had at the commencement of treatment might have possessed a VDR:AR of, as an example, 15:14 (which would become 15:5 on finasteride [very healthy hair]), but in the X years of treatment, while the VDR still downregulates due to inactivation, this ratio might drop to say 11:5. So the follicle produces healthy hair while on the drug but, following cessation, drops to a ratio of 11:14 and results in the hair being lost.
Explaining why VDR upregulation promotes growth or why downregulation hinders it is one thing, but explaining why the AR seems to destroy hair is another entirely. My second reason why the VDR:AR homeostasis is so important has to do with what exactly the two receptors do inside the follicles, and that can be put as simply as ‘cellular metabolism’. This was the breakthrough for me: the AR and VDR play a direct role in cellular energy production. You’ve heard it a million times before: “the mitochondria is the powerhouse of the cell”, but its importance cannot be understated.
Hair growth is a process that requires energy, cellular proliferation and DNA transcription (replication), and mitochondria provide this. A by-product of any metabolic process is oxidative waste, and the health of a cell is largely dependent on the body’s capacity to bind and remove oxidative waste from it (exactly what an anti-oxidant does), and this also contributes to the health of mitochondria. This is where my theory (in my opinion) became fully-fledged: AR has been shown to reduce mitochondrial function [25], reduce their capacity to properly replicate DNA and negatively affect oxidative processes [26]. Inversely, the VDR promotes healthy mitochondria, and “in the long run, the absence of the [VDR] caused impairment of mitochondrial integrity and, finally, cell death.” [27]. So there I had it, the reason why a naturally-produced androgen and its receptor might cause hair loss.
Further to this, it also serves to reason that this might be minoxidil’s mystery mechanism, as it seems to increase mitochondrial efficiency by mediating certain pathways at a cellular level [28] [29]. What’s interesting is that “minoxidil induced Ca2+ (calcium) influx can increase stem cell differentiation and may be a key factor in the mechanism by which minoxidil facilitates hair growth.” [29], but what’s fascinating is that “the VDR signaling system is essential in overall Ca2+ homeostasis. Acute exposure to 1,25(OH)2D3 increases the mean open time and plasma membrane Ca2+ permeability…” [30]. While this last excerpt refers to blood calcium, I think logic lends itself to the idea that if the VDR can activate calcium mobility throughout the body, then an activated receptor inside a hair follicle cell could mobilise calcium into its mitochondria. I also think this is why minoxidil hair is also commonly referred to as “zombie” hair, as the drug artificially activates follicular mitochondria through a similar mechanism as the VDR does, but without any upregulation of the receptors that keep it alive for a longer period.
So where does all this leave us? I’m not sure. It’s one thing to be sure that the VDR:AR is crucial for hair growth (and overall bodily health), but improving it safely (if at all) is a different matter. Vitamin D affects calcium homeostasis, and too much of it can result in hypercalcemia (too much calcium in the blood) and cause more problems than it ever alleviates. I think the strategy needs to be a three-pronged approach: upregulating VDR, downregulating AR – but not to a point below healthy levels, and improving mitochondrial health.
I take quite a few things for hair loss, but I think these are the main heroes: I am on 1mg daily finasteride because I believe that (unless you’re treating hair loss pre-emptively) once you see any sign of hair loss, the cascade of AR upregulation, VDR downregulation and mitochondrial damage has well and truly begun. If I’m right in my theory, then this drug only needs to be taken until results are seen and stabilise. My thinking is that if you downregulate AR in the scalp back to a healthy density (that is lesser than VDR) then you could ween yourself off of it permanently – that is, assuming you could keep a good VDR:AR solely through VDR activation.
I’ve been researching androgenetic alopecia (Androgenetic Alopecia) considerably for a few years now ever since I first noticed its effect on myself. Like many, I pussy-footed around the reality of the situation, convincing myself that the higher hairline and thinning crown was stress-related Telogen Efflivium, some basic dietary deficiency or the result of too much booze and smokes. This meant that I delayed treatment with more conventional (and proven) methods for a little longer than I probably should have.
My apprehension to begin these treatments was by enlarge because of how unsatisfied I was with the widely-accepted ‘cause’ of hair loss: that, not everybody (for some reason), but a large group (for some reason) of unlucky fellows and some women develop a bizarre, localized sensitivity (for some reason) to an androgen that their own body produces naturally and that (for some reason) this hormone ‘attacks’ tens of thousands of tiny organs on a specific part of their head (… for some reason). I thought the logic was dumb and that that kind of thinking did not a cure make.
Now, the human body is by no means a perfect machine, but it didn’t sit well with me that certain genomes would evolve something as complicated and energy-consuming as hair growth only to have it inadvertently switched off by a naturally-occurring androgen. There were also other evidences that it wasn’t just a matter of androgen activity: how often people with Androgenetic Alopecia do not display more testosterone (or its more active metabolite dihydrotestosterone [DHT]) than people unaffected; that simply blocking DHT production does not regrow hair in all people; where and the pattern in which the hair is lost (and isn’t); and lastly, the irrefutable links Androgenetic Alopecia has to issues elsewhere in the body such as heart and prostate health.
So I dug deeper into other theories a little more substantial than “DHT BAD” – ideally to find one that gave some insight into the process of hair loss being something the body decides to do rather than something it has done unto it. I read through almost every major theory on the web: tension in the galea; reduced brown adipose tissue; mast cell activation, histamine or prostaglandin imbalance etc. None of them seemed outrageous, but none of them gave much reason for Androgenetic Alopecia other than some people simply lose the genetic lottery. Eventually I stumbled upon something I did like: the phenomenon of seasonal hair growth [1]. Put simply, the idea is that the scalp is like a UV-ray sponge for the body to use to produce vitamin D, and the amount of hair on the scalp can be considered a valve that closes (grows) to reduce UV exposure, or opens (sheds) to increase it.
This theory (and the various studies relating to it) appeased me because they suggested that an intentional and strategic biological hair loss mechanism exist in every human, regardless of whether they suffer from more permanent types of alopecia. It was comforting to know that even the thickest head of hair technically undergoes ‘hair loss’ for a large portion of the year. My thinking then became that some of us must get stuck in a sort of ‘perpetual winter’ state where the valve gets stuck open.
I was obviously nowhere near the first person to theorise that vitamin D played a major role in alopecia (as a quick Google search would tell you), but the cure seemed simple: if the body has a mechanism to keep itself in a desired range of circulating cholecalciferol (vitamin D3), then oral supplementation – which has been proven to increase serum cholecalciferol [2] – might make the body think it’s getting enough sun exposure to close the valve and regrow hair for good. However, a quick skim of numerous Reddit and hair loss forum discussions quickly shoot down that idea, with many members reporting no major (although some minor) improvements from vitamin D3 supplementation alone, with bloodwork confirming they are not deficient. So it would appear we’re back to square one and pointing the finger angrily at DHT again.
However, cholecalciferol in and of itself is largely useless when it comes to hair. What is important is the vitamin D Receptor (VDR). The VDR is a nuclear receptor found in almost every cell in the body – including scalp hair follicles – and plays a direct role in the cellular proliferation, function and health of hair growth [3]. To make it clear, a follicle with more activated VDR will produce thicker hair faster than a follicle with less VDR activation.
The issue with the simplicity of my initial thinking was that cholecalciferol cannot adequately activate the VDR by itself. It must first undergo conversion to calcifediol (25-hydroxyvitamin D) in the liver, and then undergo another conversion in the kidneys to its active form, 1,25-dihydroxycholecalciferol, also known as calcitriol. Calcitriol can activate the VDR and actually has such a high affinity (roughly 1000 times greater than cholecalciferol) for doing so that the VDR is sometimes referred to as the ‘calcitriol receptor’ in medical literature.
Where it gets confusing (and more than a little frustrating), is that you can have more than one blood vitamin D value that may not correlate to the other. Most doctors will test for ‘25 OH vitamin D’, also known as 25-hydroxyvitamin D, or calcifediol, as mentioned earlier. This value is good for testing for deficiency, but is more or less useless in relation to VDR activation as calcifediol cannot activate the receptor well either. Doctors can test for 1,25-dihydroxyvitamin D (calcitriol), but this will not give an insight into the body’s stores of the fat-soluble vitamin. In fact, blood calcitriol can actually spike if there is a deficiency of stored calcifediol, which further complicates the results [4].
Deciphering this means, basically, that your common bloodwork for vitamin D can come back with healthy (or even above-ideal) numbers of stored, inactive D3, but shed zero light on the circulating calcitriol metabolite that can activate the VDR. It’s also been established that the ratio at which calcitriol is converted from calcifediol (the 1,25D:25D) can vary substantially from person to person: “the ratio between calcitriol and calcifediol serum concentrations could suggest vitamin D hydroxylation efficiency.” [5]. Hydroxylation is “a chemical process that introduces a hydroxyl group (-OH) into an organic compound. In biochemistry, hydroxylation reactions are often facilitated by enzymes called hydroxylases.” [6]. What this means is that hydroxylation is what makes regular vitamin D (from sun exposure or a supplementation) able to activate the VDR and grow hair, and that it’s possible that a lowered natural capacity for vitamin D hydroxylation could result in less circulating calcitriol and thus lower systemic VDR activation.
While I couldn’t find any studies alluding to what the 1,25D:25D ratio might have been in our forefathers, it is not hard to find studies referencing vitamin D’s hydroxylation cofactors – the primary one of which being magnesium, an element that a huge proportion of the population are deficient in and one stripped from the body by modern diet and lifestyle [7]. On top of magnesium being necessary to produce calcitriol (and thus directly implicated in VDR activation), it’s also been shown that inadequate magnesium levels lead to calcification of soft tissues and that adequate levels protect against it [8]. Interestingly, and as we probably all know by now, biopsies of bald scalps have shown considerable calcification in their soft tissues compared to non-balding scalps. More interestingly, is that the negative effects of low magnesium can also be seen in the heart and prostate (in hypertension and benign prostate hyperplasia) – and that these are the organs and conditions that the only two FDA-approved medications for hair loss (minoxidil and finasteride) were originally designed to alleviate.
So, all I’ve really talked about thus far is the VDR. The glaring hole in the theory is that the internet has seen a plethora of success stories from people taking anti-androgens and minoxidil (among others that I probably won’t get into). “Hey, idiot, if the VDR is solely responsible for hair cycling, then why do these drugs that don’t have any direct action on the VDR work for so many people?” you might ask, and it’d be a damn good question. I stalled on this part of my theory for a long time but I’m glad I did, because I think this is where mine differs from others, so here it is, in a nutshell…
I believe Androgenetic Alopecia is the result of a bad ratio of VDR to androgen receptor (AR) activation and density within the body.
There seems to me to be four primary and irrefutable facts that science has concluded regarding these receptors and their relation to hair growth and Androgenetic Alopecia:
· One: activating the AR in a hair follicle (like DHT can) causes a decrease of the anagen (growth) phase and eventually a complete miniaturisation of the follicle. [9]
· Two: activating the VDR in a hair follicle (like calcitriol can) causes an increase of the angagen phase which produces a thicker, faster-growing hair. [3]
· Three: inhibiting activation of the AR in a hair follicle (like an anti-androgen can) arrests further hair loss in most people, regrows in some but does not prevent further loss in others. [10]
· Four: inhibiting activation of the VDR in a hair follicle (like low levels of calcitriol or low VDR density can) causes a decrease of the anagen phase and eventually shedding. [11]
My theory is this: I believe there exists an ideal homeostasis of VDR and AR inside every organ in the body for healthy and proper function, but most modern diets and lifestyles are throwing out this balance. It’s important to place equal significance on each of these receptors in terms of Androgenetic Alopecia. I think of the balance as a tug-of-war, with the AR wanting to turn the follicle down to zero and the VDR wanting to turn it to max. As long as the ratio of VDR:AR tilts in the VDR’s favour by even the slightest amount, you will keep your hair.
This might also allude as to why it’s exceptionally rare, but not impossible for women to develop Androgenetic Alopecia, as they have very low (but never zero) circulating androgens.
Two quite ironic cases I came across in my research show the power of each receptor in isolation. Firstly, eunuchs (boys that are castrated before puberty and don’t ever produce considerable quantities of testosterone or DHT) never lose their hair [12], and secondly, a case of a seven year old boy with hereditary vitamin D-dependent rickets type II ([VDDR-II] (a condition where the VDR is resistant to activation from calcitriol, thus experiencing almost zero activation systemically,) who suffered alopecia totalis at an age too young to have begun developing sizeable amounts of testosterone or DHT [13]. So, we have grown men who never lost a single scalp hair because of minuscule AR receptor activation, and a young boy who never grew a single scalp hair because of miniscule VDR activation.
It was all starting to make sense to me, but I still wasn’t happy with why exactly the AR – a receptor that exists naturally in the follicle – would effectively act to damage it. With further reading, I came to the conclusion that there are two primary reasons why this homeostasis is so important and why going out of it leads to almost exponential and often irreversible loss. The first of which has to do with not only the ratio of receptors themselves, but also how they interact with the other:
While there has not been extensive research into the interplay (crosstalk) of these two receptors within hair follicles, there have been numerous studies regarding how they react in other tissues – namely the prostate. Although vastly different organs, the 5-alpha-reductase type II enzyme ([5ARII], one of three responsible for converting testosterone into DHT,) is most prevalent in scalp and prostatic tissues [14]. Finasteride has only been approved by the FDA to deal with complications in these two tissues by selectively targeting the 5ARII enzyme, suggesting there may be a similar mode of suppression in each. In studies of prostate cancer, VDR activation has been shown to suppress AR expression in certain cell lines [15] while AR activation has been shown to suppress VDR activation [16] [17].
Further to the expressional crosstalk between the two receptors, there is also the matter of up/down regulation and receptor density. Cells in the prostate and scalp possess both AR and VDR (among other receptors) in varying densities. There have been numerous studies showing that: AR can be upregulated with regular activation [18] [19]; AR can be downregulated by prolonged inactivation [20]; VDR can be upregulated (and protected) by activation [21] [22]; VDR can be downregulated by deficiency [23].
For me, this explains why there are such varying severities of hair loss amongst individuals and why anti-androgens such as finasteride work for some better than others. I believe the issue and cure is not simply reducing AR activation, but reducing it to a point where it is exceeded by VDR activation. Finasteride reduces scalp DHT by roughly 64% at a 1mg daily dosage [24]. As a very rudimentary example, say you have some hair follicles with 20 active AR but only 12 active VDR. A 1mg dose of finasteride should (in theory) reduce that number of activated AR by 64% to around 7.2, creating a surplus of VDR activation and thus regrowing hair. This is what I believe happens in follicles that respond well to anti-androgen treatment.
But say, due to VDR downregulation and AR upregulation (which often go hand-in-hand), you had follicles with 30 active AR and only 10 active VDR. That same finasteride dose will create a larger drop in AR activation than the previous example (19.2 vs 12.8), but will only bring the total activated AR down to 10.8 – still exceeding the activated VDR count and thus maintaining an environment where the VDR is under-expressed compared to the AR and hair loss continues. My theory posits a few things here: firstly, that great responders to anti-androgens do not have a major deficit in their VDR:AR to contend with; secondly, that poor responders to anti-androgens do have a major deficit in their VDR:AR that some anti-androgens are not powerful enough to correct; and thirdly, that anti-androgens such as finasteride rarely produce a complete reversal of hair loss due to differing ratios of VDR:AR across the scalp follicles, as some follicles have gone beyond the drug’s ‘saveable threshold’.
Further to this, I feel the theory also provides some explanation as to the ‘catch up hair loss’ phenomenon, whereby people who regrow hair with an anti-androgen and then end treatment often find that their hair is lost in a more severe pattern than it was pre-treatment, i.e. they lose even more hair post-treatment than they had lost before it. In these cases, I would think the healthy hair they had at the commencement of treatment might have possessed a VDR:AR of, as an example, 15:14 (which would become 15:5 on finasteride [very healthy hair]), but in the X years of treatment, while the VDR still downregulates due to inactivation, this ratio might drop to say 11:5. So the follicle produces healthy hair while on the drug but, following cessation, drops to a ratio of 11:14 and results in the hair being lost.
Explaining why VDR upregulation promotes growth or why downregulation hinders it is one thing, but explaining why the AR seems to destroy hair is another entirely. My second reason why the VDR:AR homeostasis is so important has to do with what exactly the two receptors do inside the follicles, and that can be put as simply as ‘cellular metabolism’. This was the breakthrough for me: the AR and VDR play a direct role in cellular energy production. You’ve heard it a million times before: “the mitochondria is the powerhouse of the cell”, but its importance cannot be understated.
Hair growth is a process that requires energy, cellular proliferation and DNA transcription (replication), and mitochondria provide this. A by-product of any metabolic process is oxidative waste, and the health of a cell is largely dependent on the body’s capacity to bind and remove oxidative waste from it (exactly what an anti-oxidant does), and this also contributes to the health of mitochondria. This is where my theory (in my opinion) became fully-fledged: AR has been shown to reduce mitochondrial function [25], reduce their capacity to properly replicate DNA and negatively affect oxidative processes [26]. Inversely, the VDR promotes healthy mitochondria, and “in the long run, the absence of the [VDR] caused impairment of mitochondrial integrity and, finally, cell death.” [27]. So there I had it, the reason why a naturally-produced androgen and its receptor might cause hair loss.
Further to this, it also serves to reason that this might be minoxidil’s mystery mechanism, as it seems to increase mitochondrial efficiency by mediating certain pathways at a cellular level [28] [29]. What’s interesting is that “minoxidil induced Ca2+ (calcium) influx can increase stem cell differentiation and may be a key factor in the mechanism by which minoxidil facilitates hair growth.” [29], but what’s fascinating is that “the VDR signaling system is essential in overall Ca2+ homeostasis. Acute exposure to 1,25(OH)2D3 increases the mean open time and plasma membrane Ca2+ permeability…” [30]. While this last excerpt refers to blood calcium, I think logic lends itself to the idea that if the VDR can activate calcium mobility throughout the body, then an activated receptor inside a hair follicle cell could mobilise calcium into its mitochondria. I also think this is why minoxidil hair is also commonly referred to as “zombie” hair, as the drug artificially activates follicular mitochondria through a similar mechanism as the VDR does, but without any upregulation of the receptors that keep it alive for a longer period.
So where does all this leave us? I’m not sure. It’s one thing to be sure that the VDR:AR is crucial for hair growth (and overall bodily health), but improving it safely (if at all) is a different matter. Vitamin D affects calcium homeostasis, and too much of it can result in hypercalcemia (too much calcium in the blood) and cause more problems than it ever alleviates. I think the strategy needs to be a three-pronged approach: upregulating VDR, downregulating AR – but not to a point below healthy levels, and improving mitochondrial health.
I take quite a few things for hair loss, but I think these are the main heroes: I am on 1mg daily finasteride because I believe that (unless you’re treating hair loss pre-emptively) once you see any sign of hair loss, the cascade of AR upregulation, VDR downregulation and mitochondrial damage has well and truly begun. If I’m right in my theory, then this drug only needs to be taken until results are seen and stabilise. My thinking is that if you downregulate AR in the scalp back to a healthy density (that is lesser than VDR) then you could ween yourself off of it permanently – that is, assuming you could keep a good VDR:AR solely through VDR activation.